Table of Contents
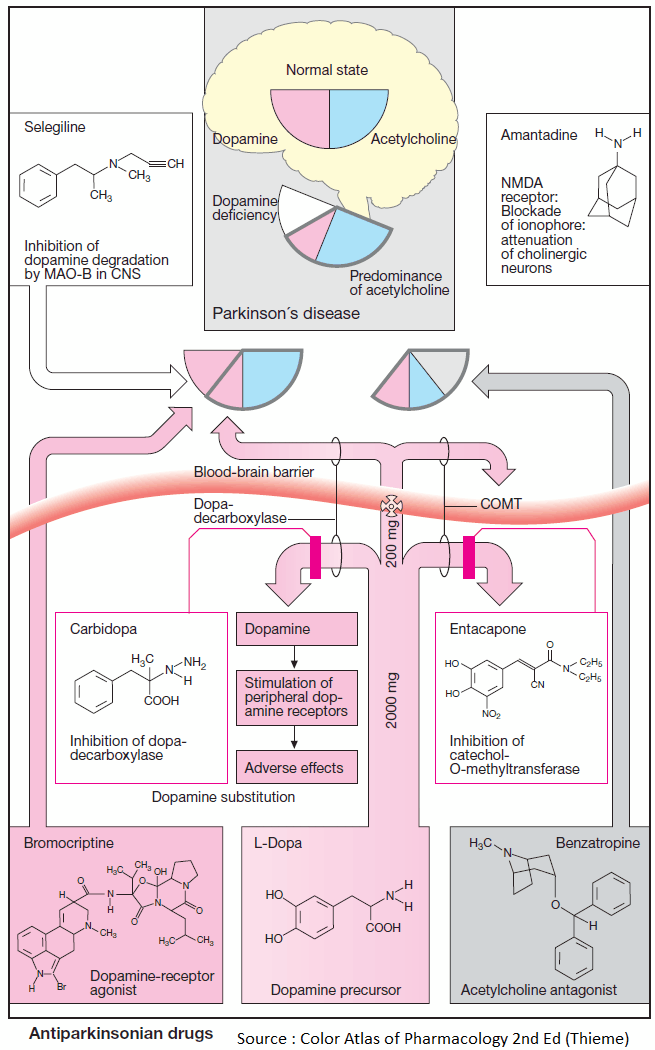
Parkinson’s disease (shaking palsy) and its syndromal forms are caused by a degeneration of nigrostriatal dopamine neurons.
The resulting striatal dopamine deficiency leads to overactivity of cholinergic interneurons and imbalance of striopallidal output pathways, manifested by:
- poverty of movement (akinesia)
- muscle stiffness (rigidity)
- tremor at rest
- postural instability
- gait disturbance
Pharmacotherapeutic measures are aimed at restoring dopaminergic function or suppressing cholinergic hyperactivity.
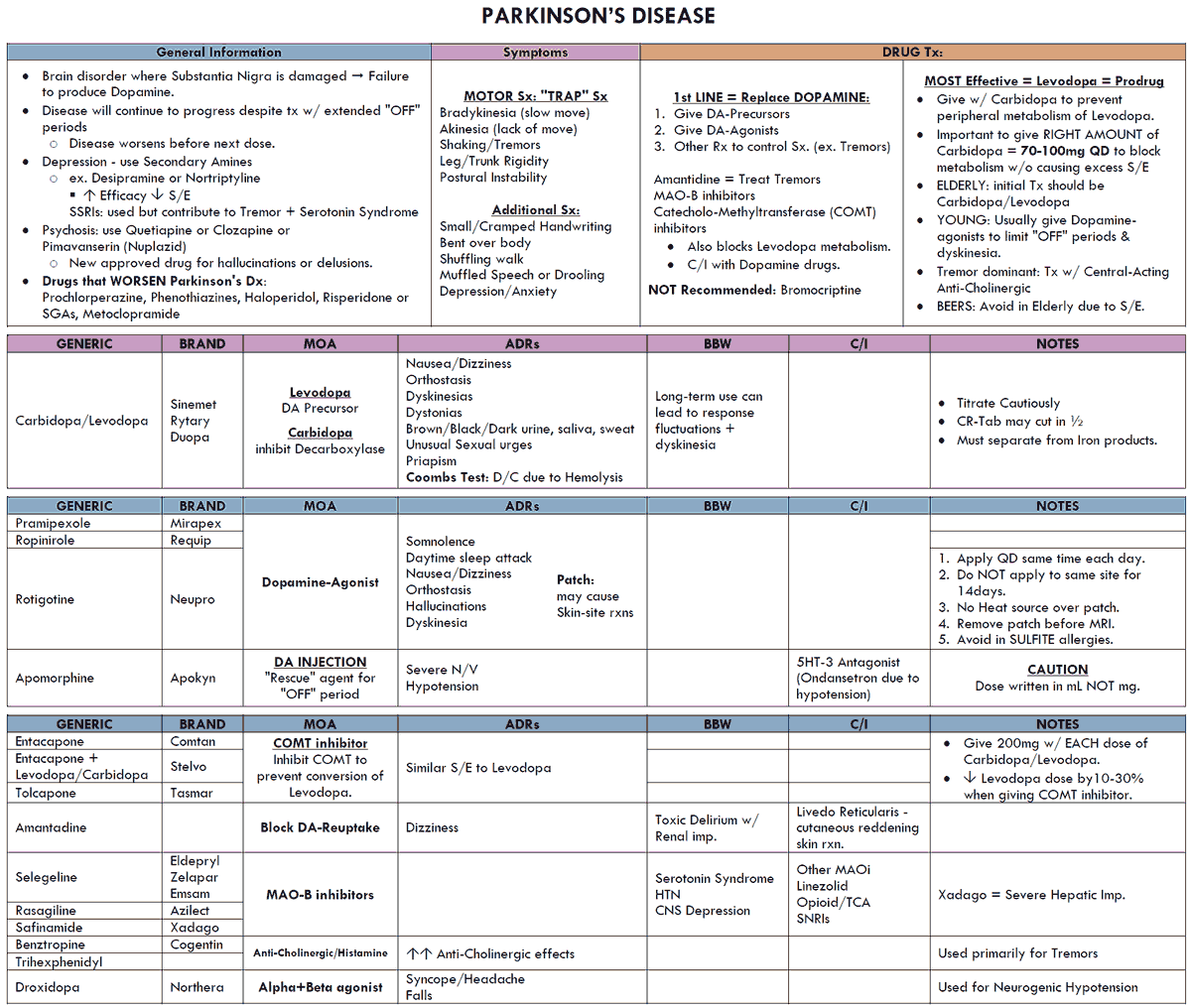
L-Dopa (Levodopa)
Dopamine itself cannot penetrate the blood-brain barrier; however, its natural precursor, L-dihydroxyphenylalanine (levodopa), is effective in replenishing striatal dopamine levels, because it is transported across the blood-brain barrier via an amino acid carrier and is subsequently decarboxylated by DOPA-decarboxylase, present in striatal tissue.
Decarboxylation also takes place in peripheral organs where dopamine is not needed, likely causing undesirable effects (tachycardia, arrhythmias resulting from activation of β1-adrenoceptors, hypotension, and vomiting).
Extracerebral production of dopamine can be prevented by inhibitors of DOPA-decarboxylase (carbidopa, benserazide) that do not penetrate the blood-brain barrier, leaving intracerebral decarboxylation unaffected.
Excessive elevation of brain dopamine levels may lead to undesirable reactions, such as involuntary movements (dyskinesias) and mental disturbances.
Dopamine receptor agonists
Deficient dopaminergic transmission in the striatum can be compensated by ergot derivatives (bromocriptin, lisuride, cabergoline, and pergolide) and nonergot compounds (ropinirole, pramipexole).
These agonists stimulate dopamine receptors (D2, D3, and D1 subtypes), have lower clinical efficacy than levodopa, and share its main adverse effects.
Inhibitors of monoamine oxidase- B (MAOB) (Selegiline)
This isoenzyme breaks down dopamine in the corpus striatum and can be selectively inhibited by selegiline. Inactivation of norepinephrine, epinephrine, and 5-HT via MAOA is unaffected.
The antiparkinsonian effects of selegiline may result from decreased dopamine inactivation (enhanced levodopa response) or from neuroprotective mechanisms (decreased oxyradical formation or blocked bioactivation of an unknown neurotoxin).
Inhibitors of catechol-O-methyltransferase (COMT) (Entacapone)
L-Dopa and dopamine become inactivated by methylation. The responsible enzyme can be blocked by entacapone, allowing higher levels of L-dopa and dopamine to be achieved in corpus striatum.
Anticholinergics
Antagonists at muscarinic cholinoceptors, such as benzatropine and biperiden, suppress striatal cholinergic overactivity and thereby relieve rigidity and tremor; however, akinesia is not reversed or is even exacerbated.
Atropine-like peripheral side effects and impairment of cognitive function limit the tolerable dosage.
Amantadine
Early or mild parkinsonian manifestations may be temporarily relieved by amantadine. The underlying mechanism of action may involve, inter alia, blockade of ligandgated ion channels of the glutamate/ NMDA subtype, ultimately leading to a diminished release of acetylcholine.
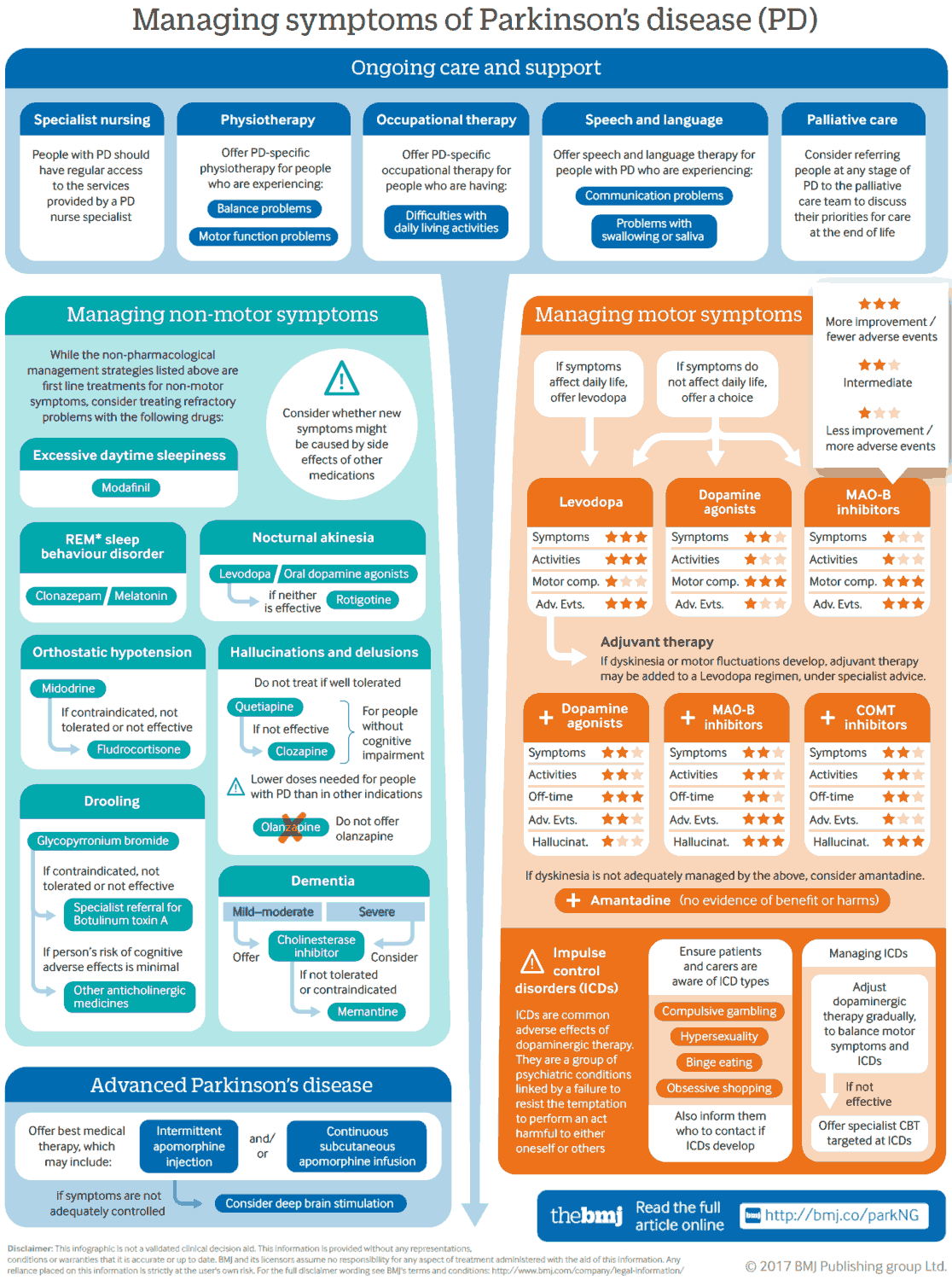
Key Points
Administration of levodopa plus carbidopa (or benserazide) remains the most effective treatment, but does not provide benefit beyond 3–5 y and is followed by gradual loss of symptom control, on-off fluctuations, and development of orobuccofacial and limb dyskinesias.
These long-term drawbacks of levodopa therapy may be delayed by early monotherapy with dopamine receptor agonists.
Treatment of advanced disease requires the combined administration of antiparkinsonian agents.