Angiotensin-converting enzyme (ACE) is a component of the antihypotensive renin-angiotensin-aldosterone (RAA) system. Renin is produced by specialized cells in the wall of the afferent arteriole of the renal glomerulus.
These cells belong to the juxtaglomerular apparatus of the nephron, the site of contact between afferent arteriole and distal tubule, and play an important part in controlling nephron function.
Stimuli eliciting release of renin are:
- a drop in renal perfusion pressure
- decreased rate of delivery of Na+ or Cl– to the distal tubules
- β-adrenoceptor-mediated sympathoactivation
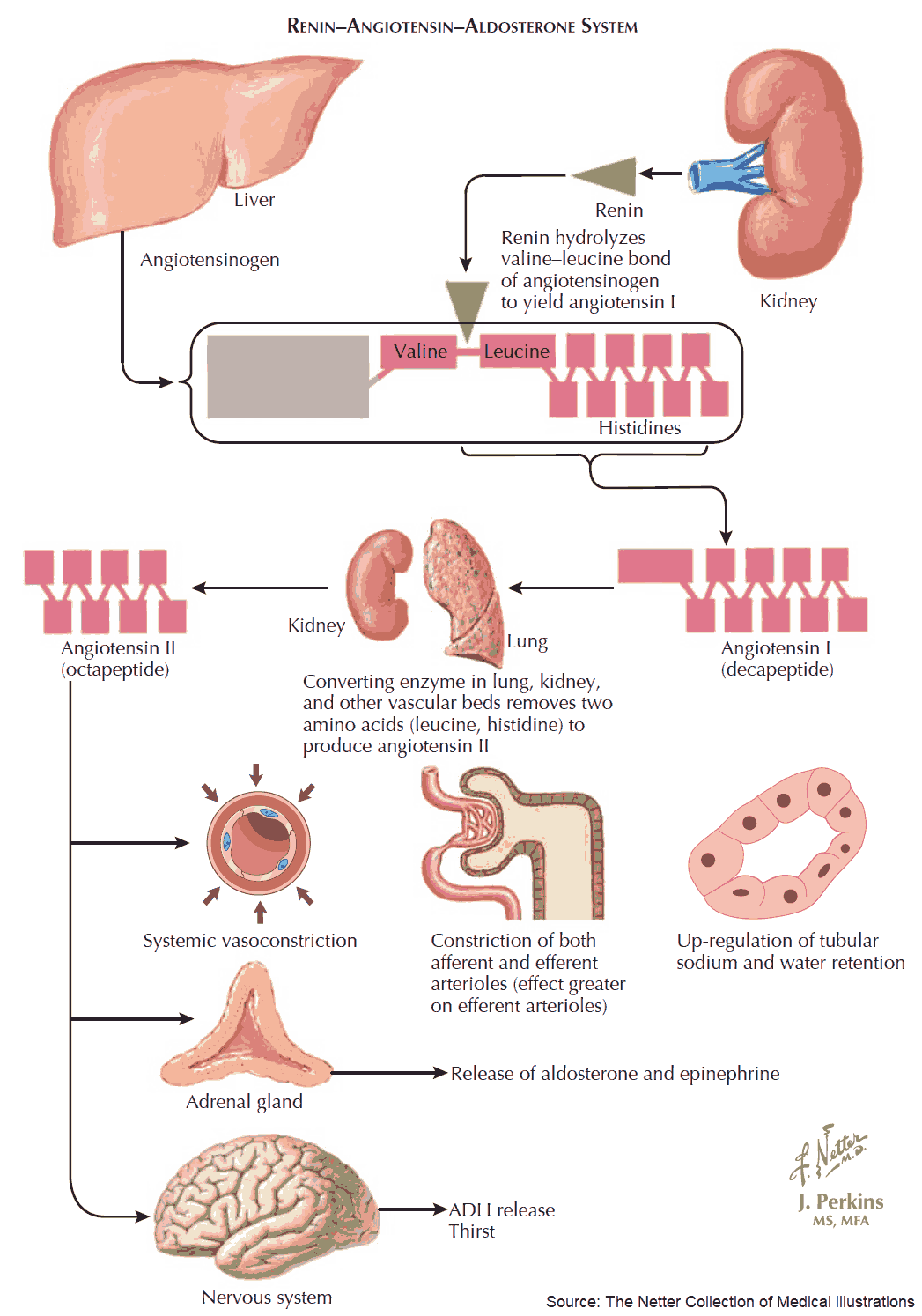
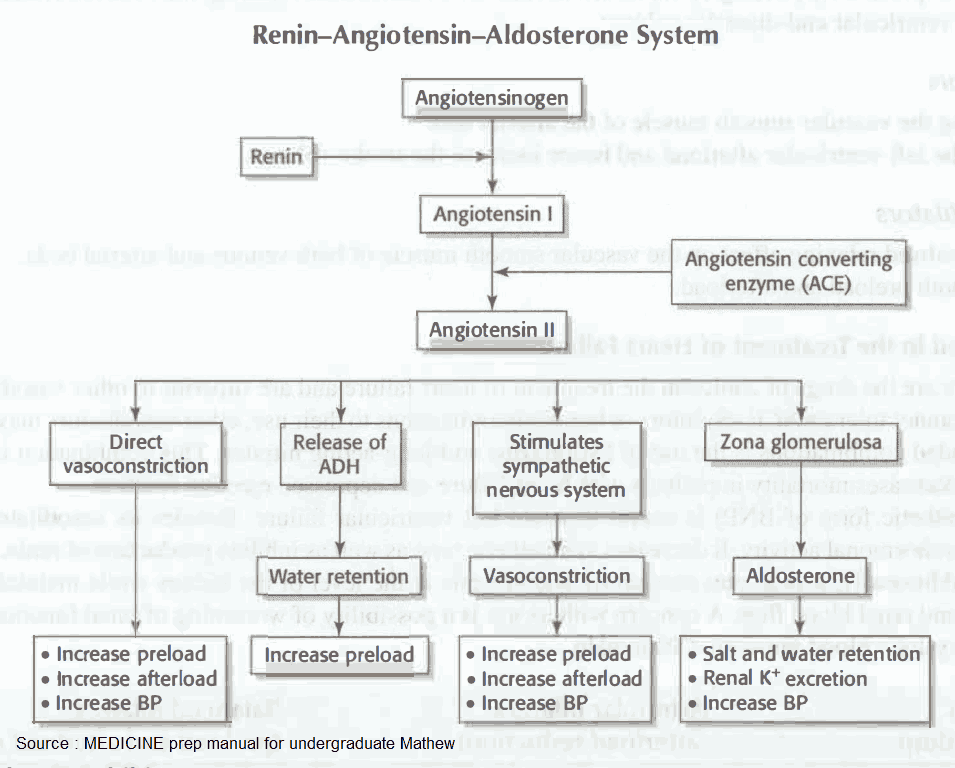
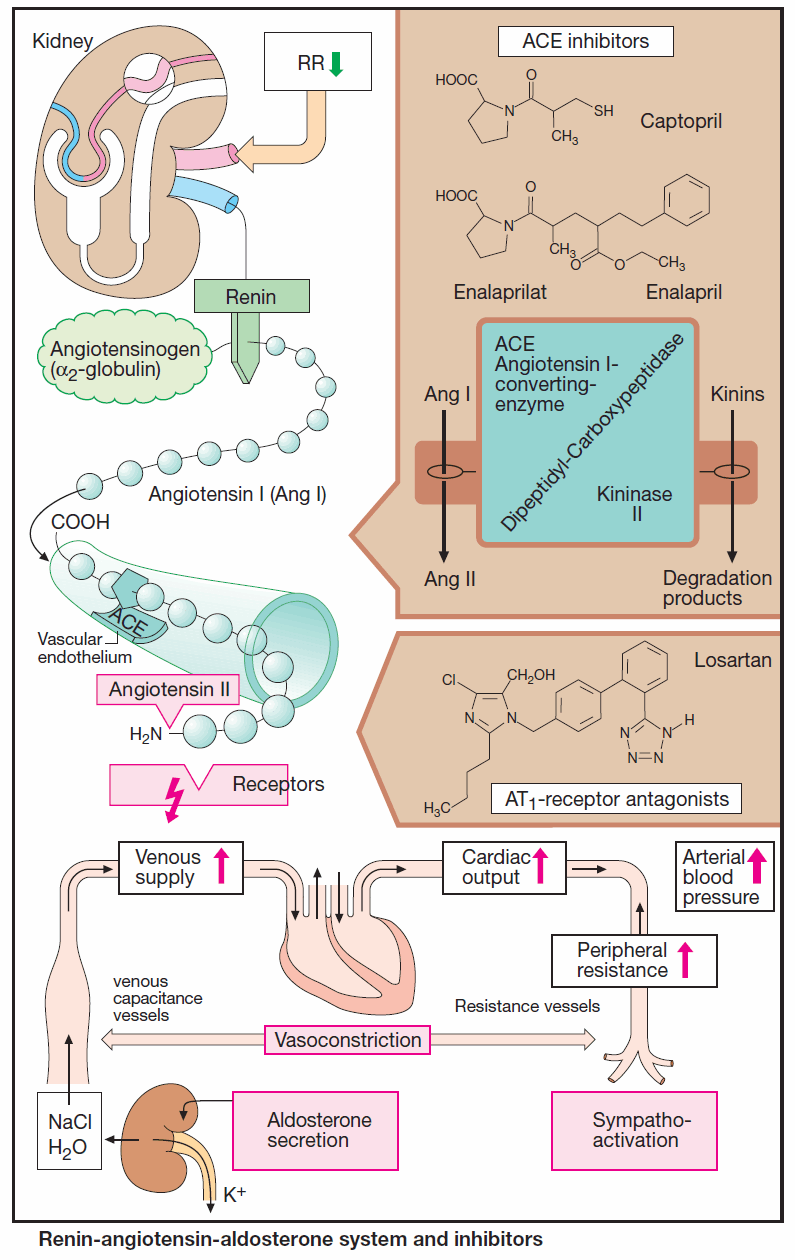
The glycoprotein renin enzymatically cleaves the decapeptide angiotensin I from its circulating precursor substrate angiotensinogen. ACE, in turn, produces biologically active angiotensin II (ANG II) from angiotensin I (ANG I).
ACE is a rather nonspecific peptidase that can cleave C-terminal dipeptides from various peptides (dipeptidyl carboxypeptidase). As “kininase II,” it contributes to the inactivation of kinins, such as bradykinin.
ACE is also present in blood plasma; however, enzyme localized in the luminal side of vascular endothelium is primarily responsible for the formation of angiotensin II. The lung is rich in ACE, but kidneys, heart, and other organs also contain the enzyme.
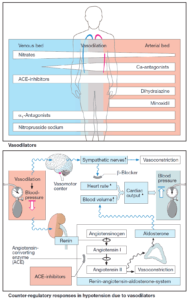
Angiotensin II can raise blood pressure in different ways, including:
- vasoconstriction in both the arterial and venous limbs of the circulation;
- stimulation of aldosterone secretion, leading to increased renal reabsorption of NaCl and water, hence an increased blood volume;
- a central increase in sympathotonus and, peripherally, enhancement of the release and effects of norepinephrine.

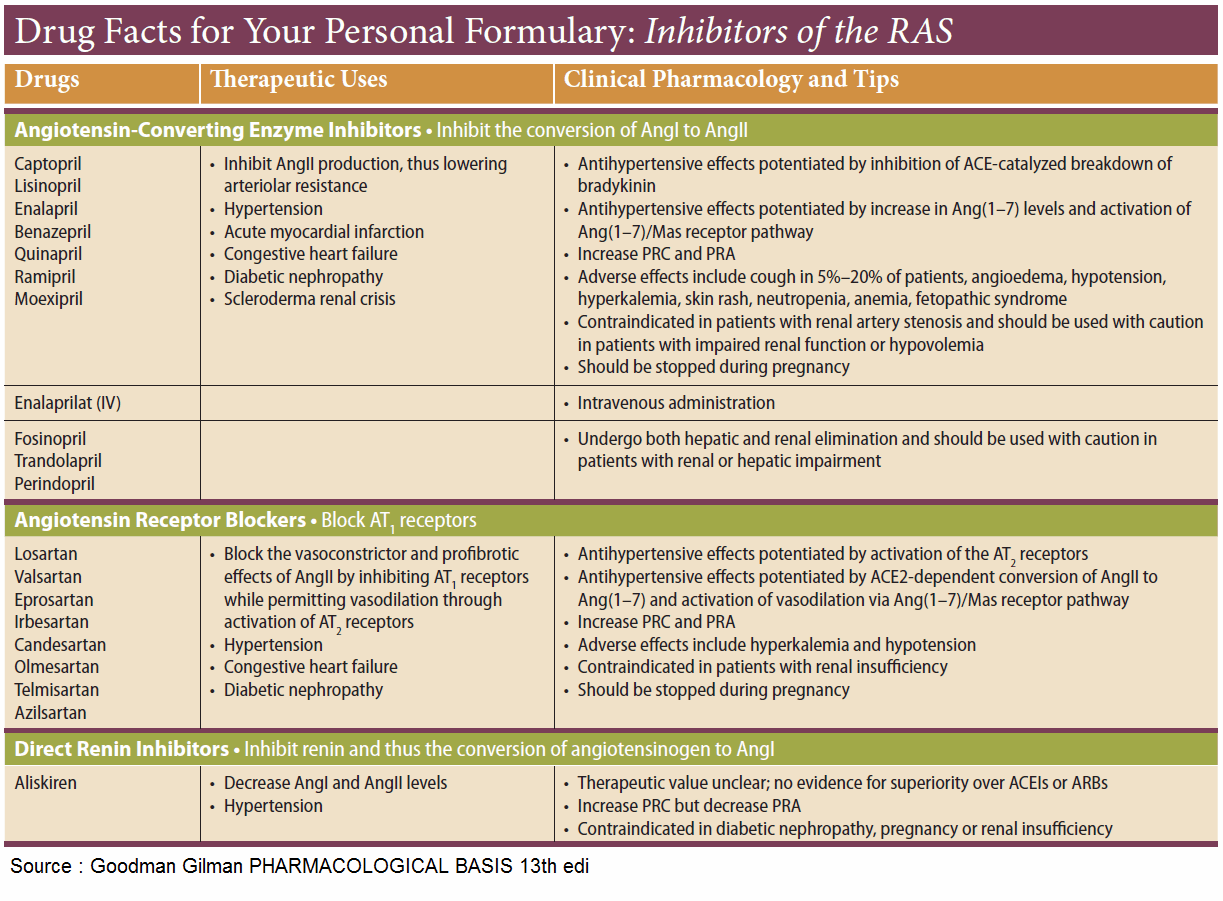
ACE inhibitors
ACE inhibitors, such as captopril and enalaprilat, the active metabolite of enalapril, occupy the enzyme as false substrates. Affinity significantly influences efficacy and rate of elimination. Enalaprilat has a stronger and longerlasting effect than does captopril.
Indications are:
- hypertension
- heart failure
- diabetic nephropathy
Lowering of an elevated blood pressure is predominantly brought about by diminished production of angiotensin II. Impaired degradation of kinins that exert vasodilating actions may contribute to the effect.
In heart failure, cardiac output rises again because ventricular afterload diminishes due to a fall in peripheral resistance. Venous congestion abates as a result of:
- increased cardiac output
- reduction in venous return (decreased aldosterone secretion, decreased tonus of venous capacitance vessels).
Undesired effects of ACE Inhibitors
The magnitude of the antihypertensive effect of ACE inhibitors depends on the functional state of the RAA system. When the latter has been activated by loss of electrolytes and water (resulting from treatment with diuretic drugs), cardiac failure, or renal arterial stenosis, administration of ACE inhibitors may initially cause an excessive fall in blood pressure.
In renal arterial stenosis, the RAA system may be needed for maintaining renal function and ACE inhibitors may precipitate renal failure.
Dry cough is a fairly frequent side effect, possibly caused by reduced inactivation of kinins in the bronchial mucosa.
Rarely, disturbances of taste sensation, exanthema, neutropenia, proteinuria, and angioneurotic edema may occur. In most cases, ACE inhibitors are well tolerated and effective.
Newer analogues include lisinopril, perindopril, ramipril, quinapril, fosinopril, benazepril, cilazapril, and trandolapril.
Antagonists at Angiotensin II Receptors
Two receptor subtypes can be distinguished: AT1, which mediates the above actions of AT II; and AT2, whose physiological role is still unclear. The sartans (candesartan, eprosartan, irbesartan, losartan, and valsartan) are AT1 antagonists that reliably lower high blood pressure. They do not inhibit degradation of kinins and cough is not a frequent side-effect.