The first QRS complex (↓) has a normal duration and is preceded by a P wave (+). This is followed by an episode of a wide QRS complex rhythm with marked variability of QRS morphology and a change in axis. This is polymorphic ventricular tachycardia.
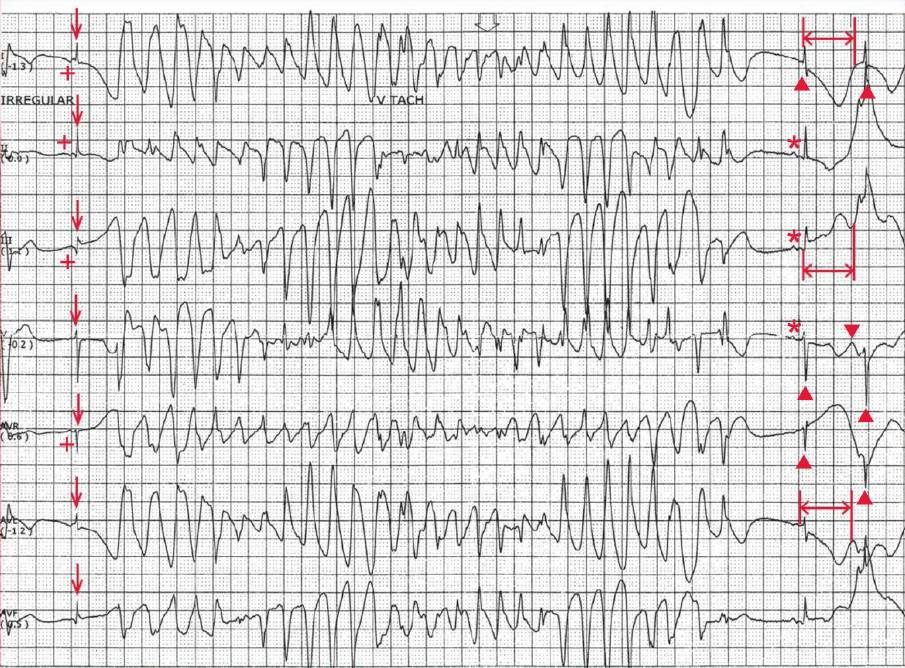
The last two QRS complexes on the ECG (▲), which are identical to the first QRS complex (↓), have a normal duration and are preceded by a P wave (*); therefore, these are supraventricular complexes, likely sinus in origin.
Although the QT interval of the first QRS complex (↓) is difficult to establish as the T wave is interrupted by the onset of the polymorphic ventricular tachycardia, the QT interval (<->) of the first narrow complex after the arrhythmia can be measured and it is prolonged (600 msec).
Therefore, the polymorphic ventricular tachycardia, which is associated with QT prolongation (of the sinus complex), is termed torsade de pointes.
Of note is that there appears to be a U wave superimposed on the T wave (▼), best seen in lead V1 (fourth line). This is referred to as a QT-U wave and is most commonly seen with congenital QT prolongation.
Along with the history of several years of seizures, which are often caused by undiagnosed torsade de pointes, this ECG pattern is typical of a congenital long QT syndrome.
Congenital Long QT Syndrome
A congenital long QT syndrome is due to a channelopathy involving a membrane potassium, sodium, or calcium channel. Although more than 10 genetic variations have been identified, the most common abnormality is a gene involving the potassium channels, resulting in the long QT (LQT1 and LQT2).
In congenital QT prolongation (ie, LQT1 or LQT2), torsade de pointes is often precipitated by exercise or an increase in sympathetic activity. With the increase in heart rate the QT interval fails to shorten appropriately and may even lengthen, increasing the risk for torsade.
In addition, sympathetic stimulation can increase the frequency and amplitude of early after-depolarizations (low amplitude membrane oscillations that occur during the prolonged phase 2 of the action potential that causes the QT prolongation), resulting in triggered activity and the occurrence of torsade.
Since sympathetic stimulation and an increase in heart rate are associated with torsade de pointes in congenital long QT syndrome, therapy with a beta-blocker is often acutely effective for preventing recurrent torsade.