Table of Contents
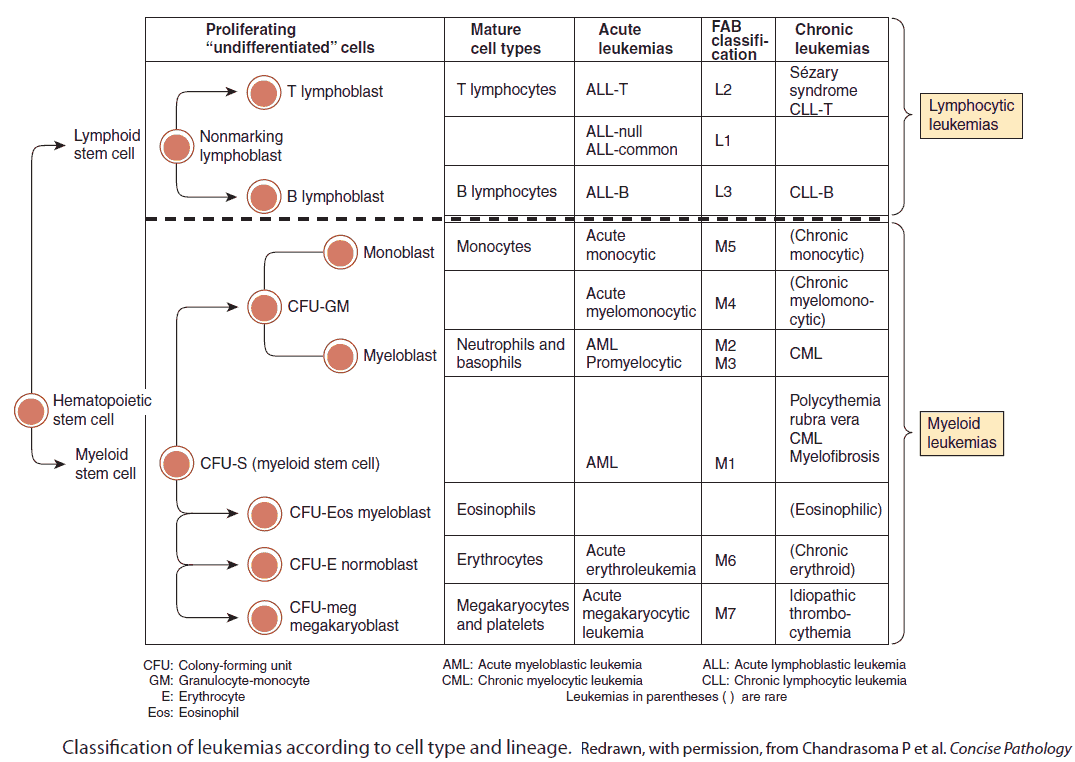
The leukemias are a group of conditions characterized by the malignant proliferation of leukocytes in the bone marrow. The cells spill out into the blood stream and may infiltrate other organs.
In the acute leukemias, there is a proliferation of early lymphoid and myeloid precursors (called blasts) which do not mature. The clinical course is very aggressive and rapidly fatal without treatment.
The chronic leukemias have a more indolent course and are characterized by the proliferation of lymphoid and myeloid cells, which reach maturity (lymphocytes and neutrophils, respectively).
Acute Lymphoblastic Leukemia
Epidemiology and etiology
Acute lymphoblastic leukemia (ALL) is the second most common malignancy in children under 15, accounting for 30% of all childhood cancers. Each year there are between 2500 and 3500 cases of ALL in the U.S. Its etiology is unknown but is probably multifactorial. Known factors include radiation and benzene. In addition, children with Down’s syndrome are at increased risk for ALL.
Pathology
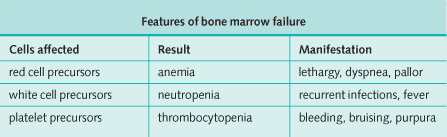
- Lymphoblasts accumulate in the bone marrow and can cause bone marrow failure.
- Lymphoblasts circulate in the blood stream and can infiltrate the lymph nodes, liver, spleen, kidneys, testicles, and central nervous system (CNS).
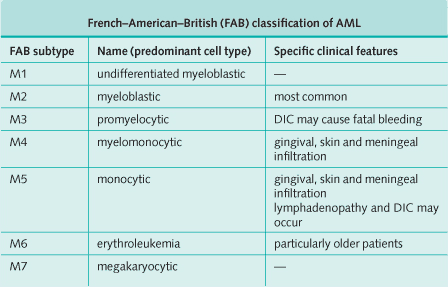
- Classified according to the French-American-British (FAB) system

Presentation
- Peak incidences at age 4 and over 65 years.
- The history is usually short as the disease is so aggressive (days to a few weeks).
- Symptoms are due to rapidly expanding tumor cells in the bone marrow, causing bone pain or bone marrow failure
- Symptoms on examination include fever, pallor, headache, thrombocytopenia (with increased risk of bleeding and ecchymosis), lymphadenopathy, and hepatosplenomegaly.
- Main sites of relapse are bone marrow, CNS, and testes.
Investigations
- Normochromic normocytic anemia with low reticulocyte count.
- High white cell count due to lymphoblasts; neutropenia may be present.
- Thrombocytopenia: 25% of patients have severe thrombocytopenia (<20,000 μL) at diagnosis.
- Bone marrow is hypercellular and dominated by lymphoblasts (usually >50%).
- Cytogenetic abnormalities may be present (e.g. hyperdiploidy or the Philadelphia chromosome [Ph]).
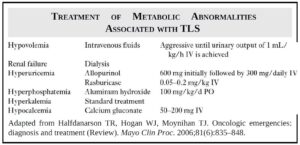
- Urate high.
- LDH high.
- Calcium, potassium, and phosphate may also be raised.
- Mediastinal mass on chest x-ray in T cell-ALL.
- Cerebrospinal fluid examination may show lymphoblasts, increased pressure, and increased protein.
Treatment
Care should be managed in a specialized unit and includes:
- Supportive care with antibiotics, blood products, and colony-stimulating factors (e.g., G-CSF for neutropenia).
- Allopurinol to prevent tumor lysis syndrome (uric acid nephropathy).
- Cytotoxic chemotherapy (induction, consolidation, maintenance. ALL treatment consists of three areas: induction of remission, consolidation, and maintenance.
- Induction of remission is achieved by using the triad of vincristine, prednisone, and anthracycline (not, however, that many different regimens are in use). Exact combinations depend on factors such as age.
- Consolidation therapy is done to eradicate any subclinical residual disease during hematologic remission. Without consolidation chemotherapy, patients with ALL usually relapse. This stage of treatment lasts 2-3 years. The numerous consolidation regimens vary in exact duration of therapy and combination of medications used. One regimen includes methotrexate biweekly and 6-mercaptopurine daily.
- Maintenance therapy is the standard of care for ALL patients of all ages despite the fact that benefit from such treatment has not been demonstrated in adults. The standard maintenance therapy includes 6-mercaptropurine and methotrexate with vincristine and prednisone, given monthly for 12-36 months.
- CNS prophylaxis (secondary to the propensity for relapse due to CNS sources): cranial irradiation, intrathecal methotrexate.
- Testes: no prophylaxis but often a site of relapse.
- Treat with radiotherapy if relapse occurs.
- Bone marrow transplantation (allogeneic or autonomous) can be curative; it is used in poor risk patients. Transplantation is done during the consolidation phase of therapy with the hope that the pretransplant myeloablative chemotherapy used, with or without whole body irradiation, will eradicate any remaining tumor cells. Currently bone marrow transplantation is recommended during the second remission after a first relapse.
Prognosis
- Children: 60% 5-year survival rate.
- Adults: 30% 5-year survival rate.
- Poor prognostic indicators include increasing age, increasing white cell count, null-cell or T-cell phenotype, male sex, and abnormal karyotype.
Acute Myeloid Leukemia and Acute Nonlymphocytic Leukemia
Epidemiology
Acute myeloid leukemia (AML) and acute nonlymphocytic leukemia (ANLL) account for 20% of all leukemias and 85% of adult leukemias.
Etiology
- Genetic predisposition: concordance in twins, Down’s syndrome, Bloom’s syndrome, Fanconi’s anemia, and ataxia telangiectasia.
- Ionizing radiation: survivors of Hiroshima and military personnel involved in nuclear test explosions.
- Chemical exposure: leather and rubber workers (benzene) or cigarette smokers.
- Previous chemotherapy: alkylating agents.
- Other drugs: chloroquine, LSD.
- Predisposing diseases: myeloproliferative diseases, multiple myeloma, aplastic anemia, myelodysplasia.
Pathology
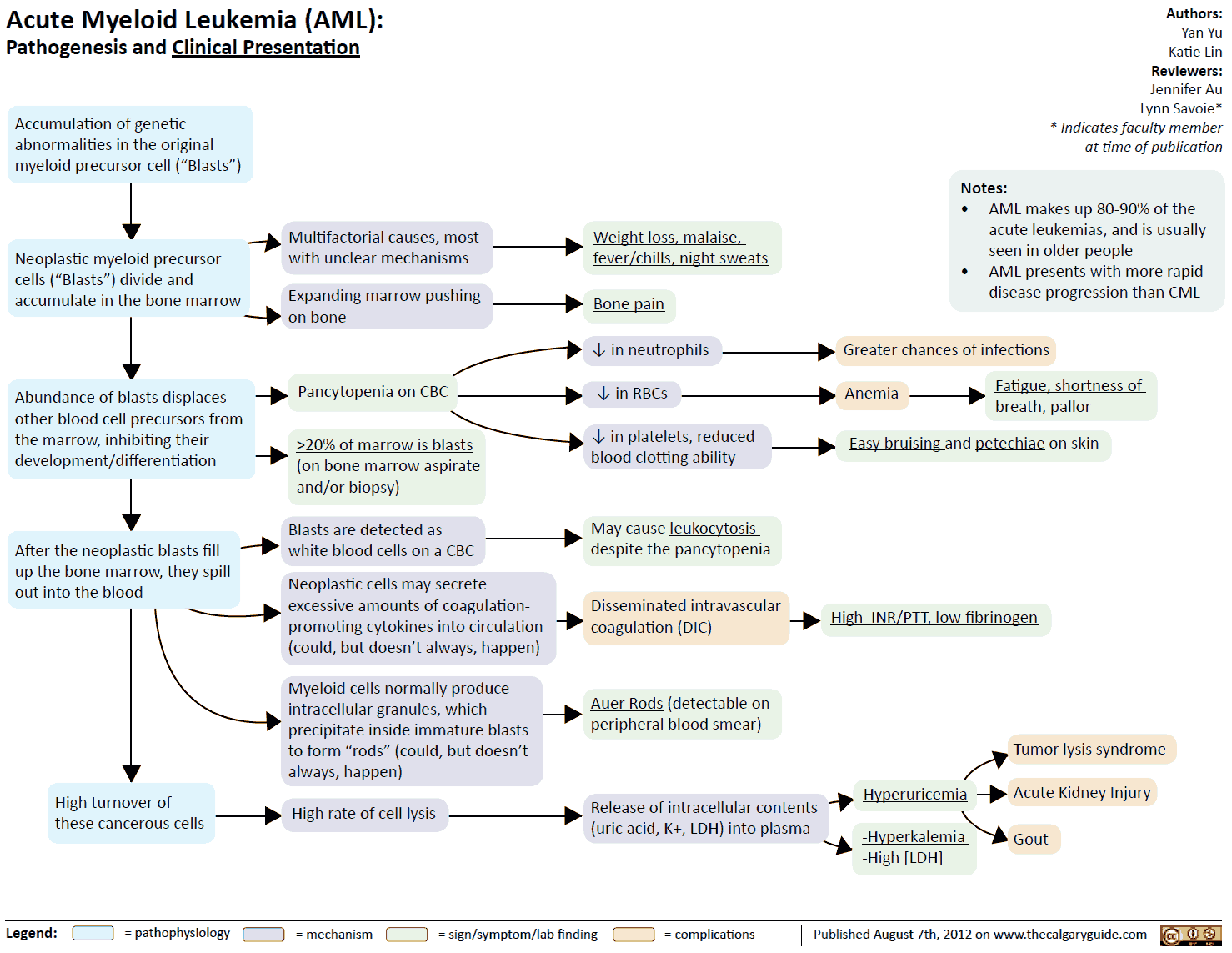
- Accumulation of immature hemopoietic cells in the bone marrow, which can cause bone marrow failure.
- Blasts can infiltrate the gums, liver, spleen, skin, and, less commonly, the CNS.
Presentation
- More frequent with increasing age (median age at presentation is 50).
- Symptoms are due to marrow failure (particularly skin infections), infiltration by leukemic cells, or bleeding (epistaxis, ecchymoses gingival bleeding).
- Bone pain, joint pain, and malaise may be prominent symptoms.
- Hepatomegaly and moderate splenomegaly are common.
- Lymphadenopathy is rare except in subtype M5.
- Gingival hypertrophy and skin lesions are features of subtypes M4 and M5.
- When the circulating white cell count is very high, leukostasis may occur, resulting in a cerebrovascular event or acute pulmonary deterioration.
Investigations
- Normochromic normocytic anemia with low reticulocyte count.
- High white cell count due to circulating blasts; neutropenia may be present.
- Blasts may contain Auer rods.
- Thrombocytopenia.
- Bone marrow is hypercellular and normal marrow is replaced by blast cells.
- Cytogenetic abnormalities may be present in around 50% of patients.
- Urate high.
- LDH high.
- Hypokalemia may occur.
- Calcium and phosphate may also be raised.
- Lumbar puncture for patients with CNS symptoms.
Treatment
Treatment should be managed in a specialized unit and includes:
- Supportive care with antibiotics and blood products.
- Allopurinol to prevent tumor lysis syndrome and gout.
- Intensive cytotoxic chemotherapy: induction, remission, and consolidation. Induction is most often a combination of cytarabine for 7 days and daunorubicin for 3 days. Consolidation therapy normally uses the same medications as induction therapy, given as 2 cycles of both duanorubicin (2 days per cycle) and cytarabine (5 days per cycle).
- Bone marrow transplantation in some patients. Bone marrow transplantation is recommended during the first remission for patients with AML. While the cure rate with transplantation is 45-65%, many of these patients would have been cured by chemotherapy alone. However, since the survival rate after bone marrow transplantation during a relapase of AML is 35% or less, it is more advantageous to undergo the procedure during the first remission.
Prognosis
- 25% long-term survivors (may be improved with transplantation).
- Poor prognostic factors include increasing age, very high white cell count, secondary leukemia (e.g., previous myelodysplasia), cytogenetic abnormalities, and the presence of disseminated intravascular coagulation (DIC).
Chronic Lymphocytic Leukemia
Epidemiology and etiology
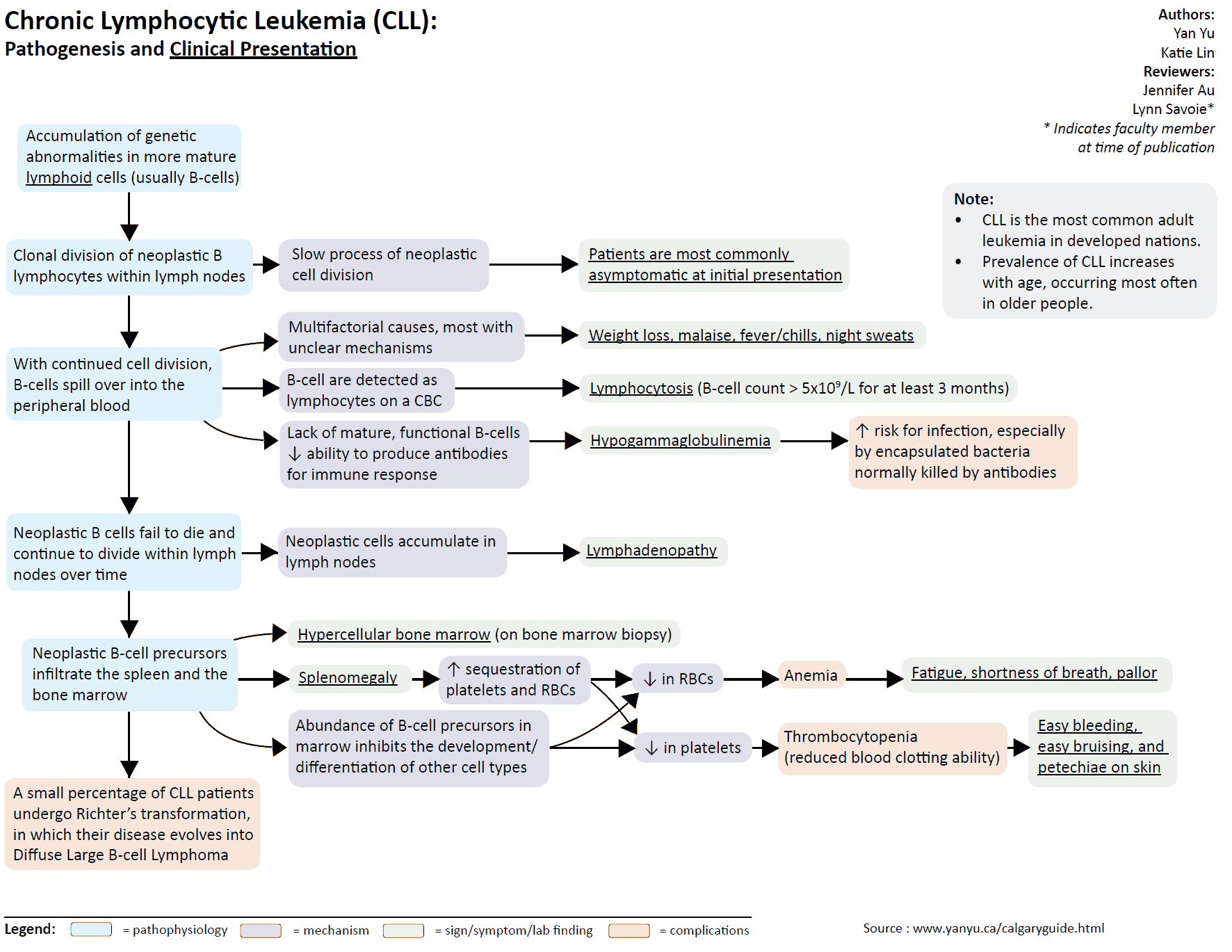
Chronic lymphocytic leukemia (CLL) is the most common leukemia in the Western Hemisphere (30% of all leukemias). Its etiology is unknown.
Pathology
- Proliferation of small lymphocytes in bone marrow, blood, and lymphoid tissues.
- These are morphologically mature but functionally abnormal.
- 95-98% of CLL patients have B-cell phenotype (remainder are T cells).
Incidence and presentation
- The most common type of leukemia in the Western World.
- Uncommon in the Far East.
- Increasing incidence with age (90% cases >50 years).
- More common in males.
- Some patients are asymptomatic; others may describe malaise, weight loss, night sweats, recurrent infections, bleeding, or symptoms of anemia.
- Lymphadenopathy is usually found (60%).
- Hepatosplenomegaly may also be present.
Investigations
- Monoclonal lymphocytosis with “smear” or “smudge” cells seen on film.
- Anemia may be due to marrow infiltration or autoimmune hemolysis (Coombs positive).
- Thrombocytopenia may be due to marrow infiltration or autoimmune destruction.
- Bone marrow shows accumulation of mature lymphocytes.
- Hypogammaglobulinemia (monoclonal gammopathy may occur).
- Lymph node biopsy: will show mature-looking lymphocytic infiltrate.
Treatment
- Observe only if asymptomatic mild disease. Currently no data suggest that early treatment increases long-term survival. Current indications to treat include anemia, thrombocytopenia, night sweats, weight loss, painful lymphadenopathy, fever, autoimmune hemolytic anemia, increasing lymphocytosis, and enlarged liver or spleen.
- Oral alkylating agents (e.g., chlorambucil or cyclophosphamide) – if there is symptomatic and progressive disease
- Intravenous fludarabine (antimetabolite) can be used in disease resistant to alkylating agents.
- Oral prednisolone for autoimmune phenomena
- Radiotherapy may be beneficial in symptomatic, localized disease.
- Splenectomy is sometimes used in refractory hypersplenism.
- Anti-CD20+ monoclonal antibody (rituximab) can also be used for both untreated and resistant disease.
- Anti-CD52 monoclonal antibody (alemtuzumab) can be used for post-remission therapy or resistant disease.
Prognosis
- Dependent on extent of disease (survival ranges from 1.5 years to over 12 years).
- In around 10% of CLL, Richter transformation to high-grade lymphoma occurs as a terminal event.
Chronic Myeloid Leukemia
Incidence and etiology
The incidence of CML is 1-2 per 100,000 (20% of all leukemias). It can be due to ionizing radiation in some patients, but often the cause is unknown.
Pathology
Malignant proliferation of myeloid cells.
Presentation
- Presents in middle age, most commonly between 40 and 60 years, with a male preponderance. Up to 50% of patients are asymptomatic at presentation.
- Symptoms include lethargy, weight loss, sweats, and left hypochondrial discomfort (enlarging spleen).
- Symptoms of anemia or thrombocytopenia may be present.
- On examination there is splenomegaly, which may be massive.
- Hepatomegaly is present in 50% of cases, but lymphadenopathy is uncommon.
- The natural history is characterized by a chronic phase lasting several years, followed by an acute, aggressive phase (the blast phase, which occurs within 3-5 years of diagnosis).
- Leukostasis may occur in the acute phase.
Investigations
- High white cell count with full range of immature and mature myeloid cells.
- Anemia may be due to marrow infiltration or hypersplenism.
- Thrombocytosis is common.
- Bone marrow demonstrates accumulation of myeloid cells.
- 95% of patients have a chromosomal translocation called the Philadelphia chromosome (Ph), which is a reciprocal translocation between chromosomes 9 and 22, producing the chimeric bcr/abl gene. Ph can be detected by FISH.
- Neutrophil alkaline phosphatase is low.
- Urate is high.
- LDH is high.
- Serum vitamin B12 is high.
Treatment
- Allopurinol orally.
- Cytotoxic chemotherapy with hydroxyurea (drug of choice) or busulphan.
- α-Interferon achieves good hematologic control and reduction in the presence of Ph.
- Allogeneic bone marrow transplantation may be curative and should be considered in young patients with an HLA-matched sibling.
- Leukopharesis will reduce the white count quickly in leukostasis.
- A new approach to treating CML has come from the understanding of the function of the Philadelphia chromosome. Given that this mutation causes the production of a novel protein via tyrosine kinase, a product was developed that can inhibit this function. ST1-571 (Gleevec) is a tyrosine kinase inhibitor that blocks the function of the bcr/abl gene fusion. Gleevec has been approved for use in CML patients with myeloid blast crisis, those who are in an accelerated phase, those who are in a chronic phase after failure of treatment, and those who have yet to receive any treatment for CML. Gleevec has been shown to be better than interferon therapy plus cytarabine for all endpoints at 18 months and should be considered the first-line therapy. Side effects include nausea, diarrhea, muscle cramp, and skin rash.
Prognosis
- Chronic phase: median time 2-6 years.
- Acute phase: median survival <3 months.
- Transformation is to AML in two-thirds of patients and to ALL in the remainder.