
Table of Contents
Coumarin Derivatives
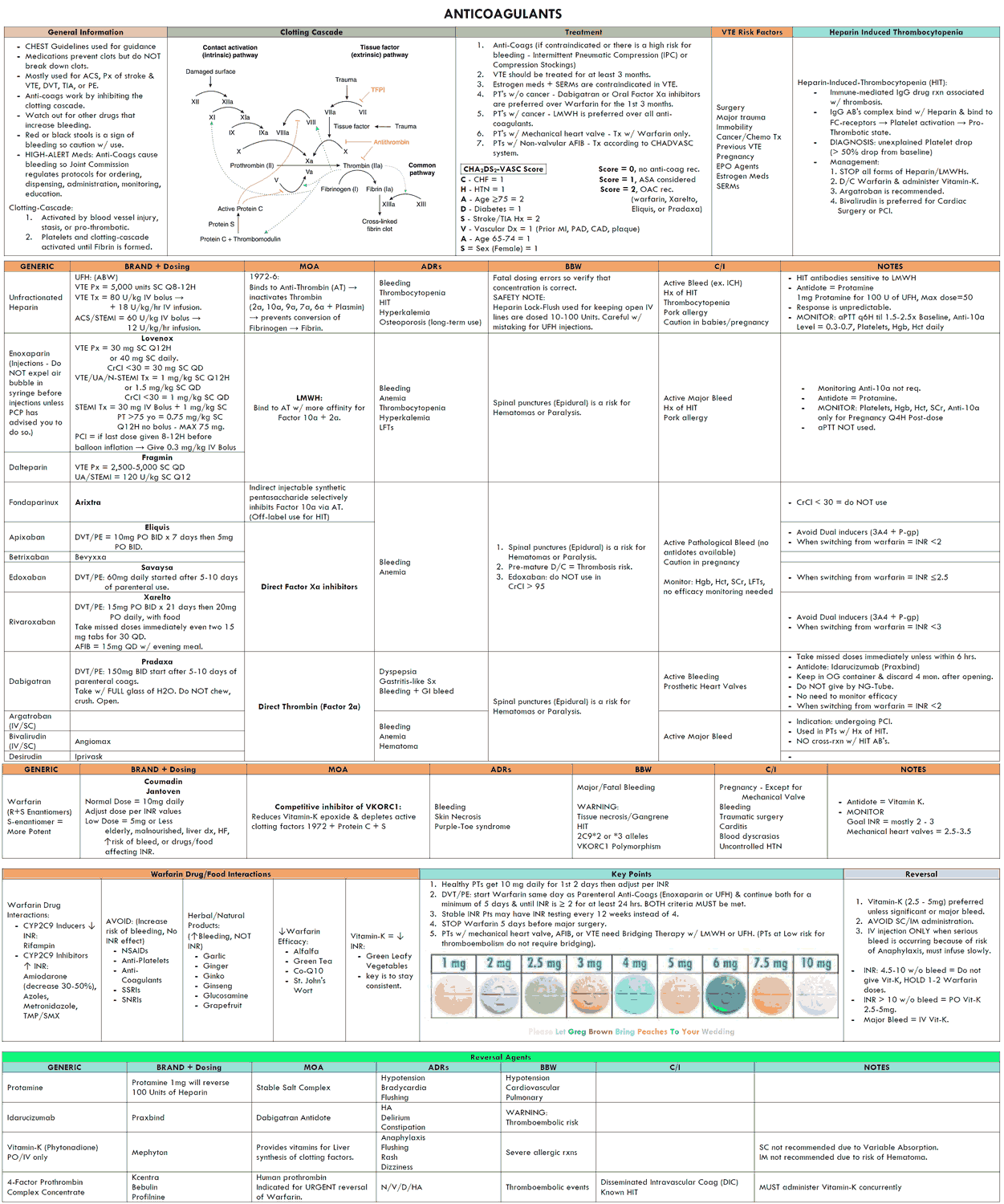
Vitamin K promotes the hepatic γ-carboxylation of glutamate residues on the precursors of factors II, VII, IX, and X, as well as that of other proteins, e.g., protein C, protein S, or osteocalcin. Carboxyl groups are required for Ca2+-mediated binding to phospholipid surfaces.
There are several vitamin K derivatives of different origins: K1 (phytomenadione) from chlorophyllous plants; K2 from gut bacteria; and K3 (menadione) synthesized chemically. All are hydrophobic and require bile acids for absorption.
Oral anticoagulants
Structurally related to vitamin K, 4-hydroxycoumarins act as “false” vitamin K and prevent regeneration of reduced (active) vitamin K from vitamin K epoxide, hence the synthesis of vitamin K-dependent clotting factors.
Coumarins are well absorbed after oral administration. Their duration of action varies considerably. Synthesis of clotting factors depends on the intrahepatocytic concentration ratio of coumarins to vitamin K.
The dose required for an adequate anticoagulant effect must be determined individually for each patient (one-stage prothrombin time). Subsequently, the patient must avoid changing dietary consumption of green vegetables (alteration in vitamin K levels), refrain from taking additional drugs likely to affect absorption or elimination of coumarins (alteration in coumarin levels), and not risk inhibiting platelet function by ingesting acetylsalicylic acid.
Adverse effects of Coumarins
The most important adverse effect is bleeding. With coumarins, this can be counteracted by giving vitamin K1. Coagulability of blood returns to normal only after hours or days, when the liver has resumed synthesis and restored sufficient blood levels of clotting factors. In urgent cases, deficient factors must be replenished directly (e.g., by transfusion of whole blood or of prothrombin concentrate).
Another side effect of Coumarins is skin necrosis, which is rare.
Read More About :
Heparin
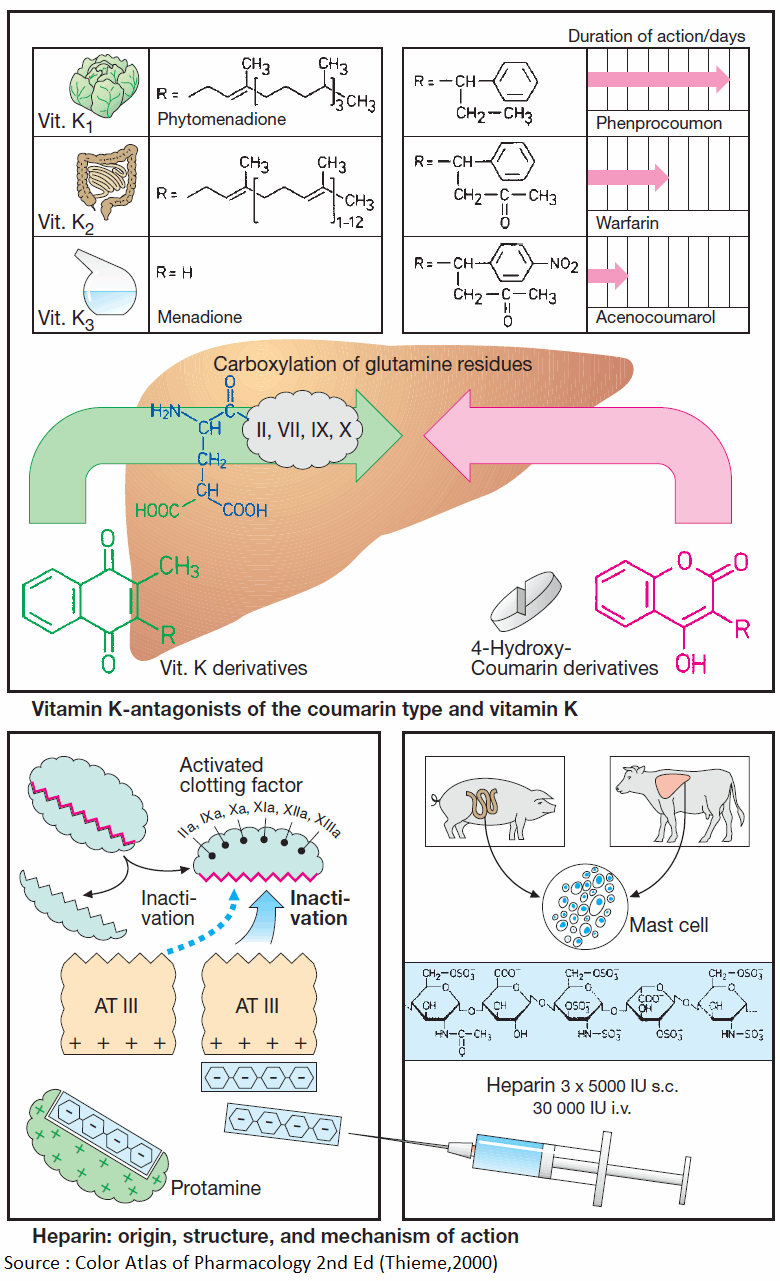
A clotting factor is activated when the factor that precedes it in the clotting cascade splits off a protein fragment and thereby exposes an enzymatic center. The latter can again be inactivated physiologically by complexing with antithrombin III (AT III), a circulating glycoprotein.
Heparin acts to inhibit clotting by accelerating formation of this complex more than 1000-fold. Heparin is present (together with histamine) in the vesicles of mast cells; its physiological role is unclear. Therapeutically used heparin is obtained from porcine gut or bovine lung. Heparin molecules are chains of amino sugars bearing -COO– and -SO4 groups.
Anticoagulant efficacy varies with chain length. The potency of a preparation is standardized in international units of activity (IU) by bioassay and comparison with a reference preparation.
The numerous negative charges are significant in several respects: (1) they contribute to the poor membrane penetrability— heparin is ineffective when applied by the oral route or topically onto the skin and must be injected; (2) attraction to positively charged lysine residues is involved in complex formation with ATIII; (3) they permit binding of heparin to its antidote, protamine (polycationic protein from salmon sperm).
If protamine is given in heparin-induced bleeding, the effect of heparin is immediately reversed. For effective thromboprophylaxis, a low dose of 5000 IU is injected s.c. two to three times daily. With low dosage of heparin, the risk of bleeding is sufficiently small to allow the first injection to be given as early as 2 h prior to surgery. Higher daily i.v. doses are required to prevent growth of clots.
Adverse effects of Heparin
- Bleeding
- Allergic reactions (e.g., thrombocytopenia)
- Heparin-induced thrombocytopenia (HIT)
- Hyperkalemia (from aldosterone suppression)
- and with chronic administration:
- reversible hair loss
- osteoporosis.
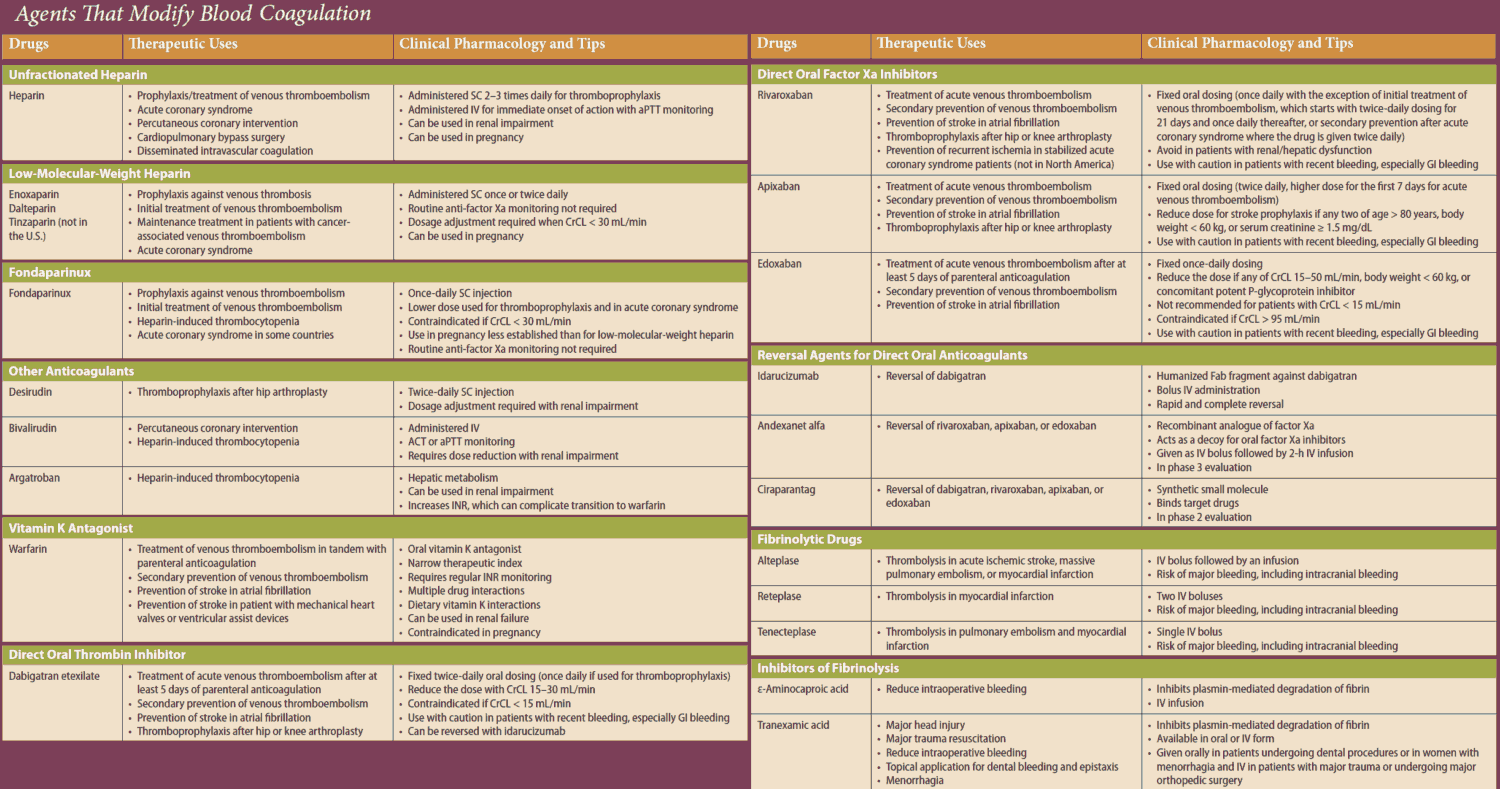
Low-molecular-weight heparin (LMWH)
Low-molecular-weight heparin (average MW ~5000) has a longer duration of action and needs to be given only once daily (e.g., certoparin, dalteparin, enoxaparin, reviparin, tinzaparin).
Frequent control of coagulability is not necessary with low molecular weight heparin and incidence of side effects (bleeding, heparin-induced thrombocytopenia) is less frequent than with unfractionated heparin.
Fibrinolytic Therapy
Fibrin is formed from fibrinogen through thrombin (factor IIa)-catalyzed proteolytic removal of two oligopeptide fragments. Individual fibrin molecules polymerize into a fibrin mesh that can be split into fragments and dissolved by plasmin.
Plasmin derives by proteolysis from an inactive precursor, plasminogen. Plasminogen activators can be infused for the purpose of dissolving clots (e.g., in myocardial infarction). Thrombolysis is not likely to be successful unless the activators can be given very soon after thrombus formation.
- Urokinase is an endogenous plasminogen activator obtained from cultured human kidney cells. Urokinase is better tolerated than is streptokinase. By itself, the latter is enzymatically inactive; only after binding to a plasminogen molecule does the complex become effective in converting plasminogen to plasmin.
- Streptokinase is produced by streptococcal bacteria, which probably accounts for the frequent adverse reactions. Streptokinase antibodies may be present as a result of prior streptococcal infections. Binding to such antibodies would neutralize streptokinase molecules.
- With alteplase, another endogenous plasminogen activator (tissue plasminogen activator, tPA) is available. With physiological concentrations this activator preferentially acts on plasminogen bound to fibrin. In concentrations needed for therapeutic fibrinolysis this preference is lost and the risk of bleeding does not differ with alteplase and streptokinase. Alteplase is rather shortlived (inactivation by complexing with plasminogen activator inhibitor, PAI) and has to be applied by infusion.
- Reteplase, however, containing only the proteolytic active part of the alteplase molecule, allows more stabile plasma levels and can be applied in form of two injections at an interval of 30 min.

Inactivation of the Fibrinolytic System
Inactivation of the fibrinolytic system can be achieved by “plasmin inhibitors,” such as ε-aminocaproic acid, p-aminomethylbenzoic acid (PAMBA), tranexamic acid, and aprotinin, which also inhibits other proteases. Lowering of blood fibrinogen concentration.
Ancrod is a constituent of the venom from a Malaysian pit viper. It enzymatically cleaves a fragment from fibrinogen, resulting in the formation of a degradation product that cannot undergo polymerization.
Reduction in blood fibrinogen level decreases the coagulability of the blood. Since fibrinogen (MW ~340 000) contributes to the viscosity of blood, an improved “fluidity” of the blood would be expected. Both effects are felt to be of benefit in the treatment of certain disorders of blood flow.
![Read more about the article All About Leukemia: From Diagnosis to Treatment [A Comprehensive Overview]](https://manualofmedicine.com/wp-content/uploads/2023/04/Classification-of-Leukemias-According-to-Cell-Type-and-Lineage-300x213.png)