Table of Contents
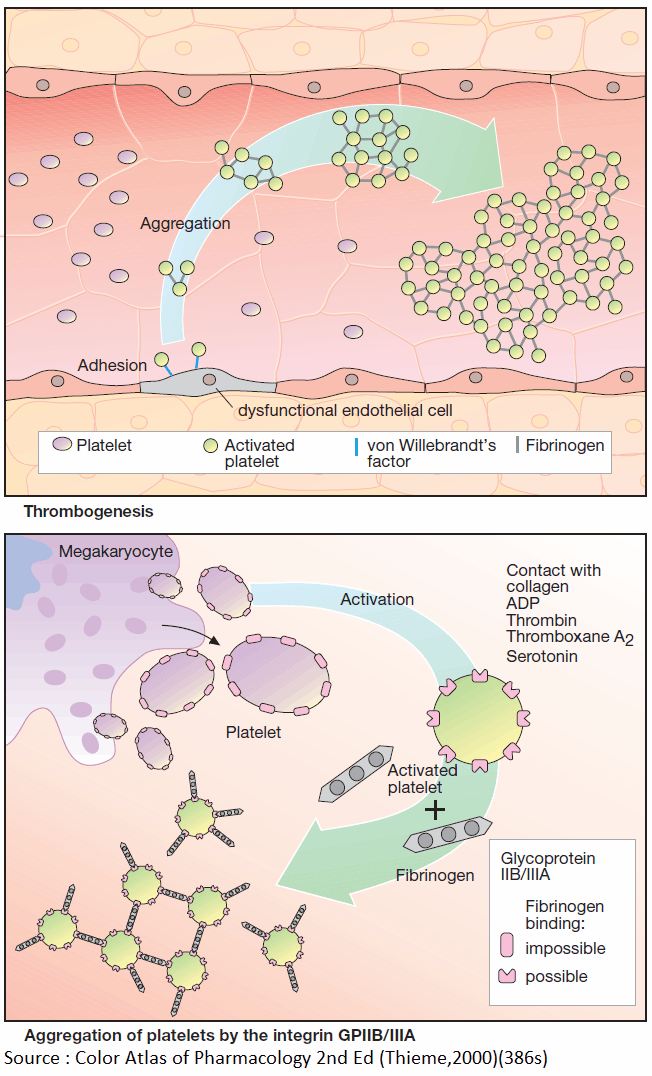
Intra-arterial Thrombus Formation
Activation of platelets, e.g., upon contact with collagen of the extracellular matrix after injury to the vascular wall, constitutes the immediate and decisive step in initiating the process of primary hemostasis, i.e., cessation of bleeding.
However, in the absence of vascular injury, platelets can be activated as a result of damage to the endothelial cell lining of blood vessels. Among the multiple functions of the endothelium, the production of NO and prostacyclin plays an important role.
Both substances inhibit the tendency of platelets to adhere to the endothelial surface (platelet adhesiveness). Impairment of endothelial function, e.g., due to chronic hypertension, cigarette smoking, chronic elevation of plasma LDL levels or of blood glucose, increases the probability of contact between platelets and endothelium.
The adhesion process involves GPIB/IX, a glycoprotein present in the platelet cell membrane and von Willebrandt’s factor, an endothelial membrane protein. Upon endothelial contact, the platelet is activated with a resultant change in shape and affinity to fibrinogen.
Platelets are linked to each other via fibrinogen bridges: they undergo aggregation. Platelet aggregation increases like an avalanche because, once activated, platelets can activate other platelets. On the injured endothelial cell, a platelet thrombus is formed, which obstructs blood flow.
Ultimately, the vascular lumen is occluded by the thrombus as the latter is solidified by a vasoconstriction produced by the release of serotonin and thromboxane A2 from the aggregated platelets. When these events occur in a larger coronary artery, the consequence is a myocardial infarction; involvement of a cerebral artery leads to stroke.
The von Willebrandt’s factor plays a key role in thrombogenesis. Lack of this factor causes thrombasthenia, a pathologically decreased platelet aggregation. Relative deficiency of the von Willebrandt’s factor can be temporarily overcome by the vasopressin anlogue desmopressin, which increases the release of available factor from storage sites.
Formation, Activation, and Aggregation of Platelets
Platelets originate by budding off from multinucleate precursor cells, the megakaryocytes. As the smallest formed element of blood (dia. 1–4 μm), they can be activated by various stimuli.
Activation entails an alteration in shape and secretion of a series of highly active substances, including serotonin, platelet activating factor (PAF), ADP, and thromboxane A2. In turn, all of these can activate other platelets, which explains the explosive nature of the process.
The primary consequence of activation is a conformational change of an integrin present in the platelet membrane, namely, GPIIB/IIIA. In its active conformation, GPIIB/IIIA shows high affinity for fibrinogen; each platelet contains up to 50,000 copies.
The high plasma concentration of fibrinogen and the high density of integrins in the platelet membrane permit rapid cross-linking of platelets and formation of a platelet plug.
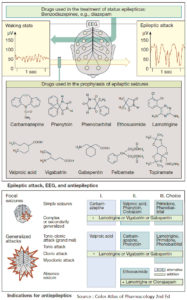
Inhibitors of Platelet Aggregation (Antiplatelets)
Platelets can be activated by mechanical and diverse chemical stimuli, some of which, e.g., thromboxane A2, thrombin, serotonin, and PAF, act via receptors on the platelet membrane. These receptors are coupled to Gq proteins that mediate activation of phospholipase C and hence a rise in cytosolic Ca2+ concentration. Among other responses, this rise in Ca2+ triggers a conformational change in GPIIB/IIIA, which is thereby converted to its fibrinogen-binding form.
In contrast, ADP activates platelets by inhibiting adenylyl cyclase, thus causing internal cAMP levels to decrease. High cAMP levels would stabilize the platelet in its inactive state. Formally, the two messenger substances, Ca2+ and cAMP, can be considered functional antagonists.
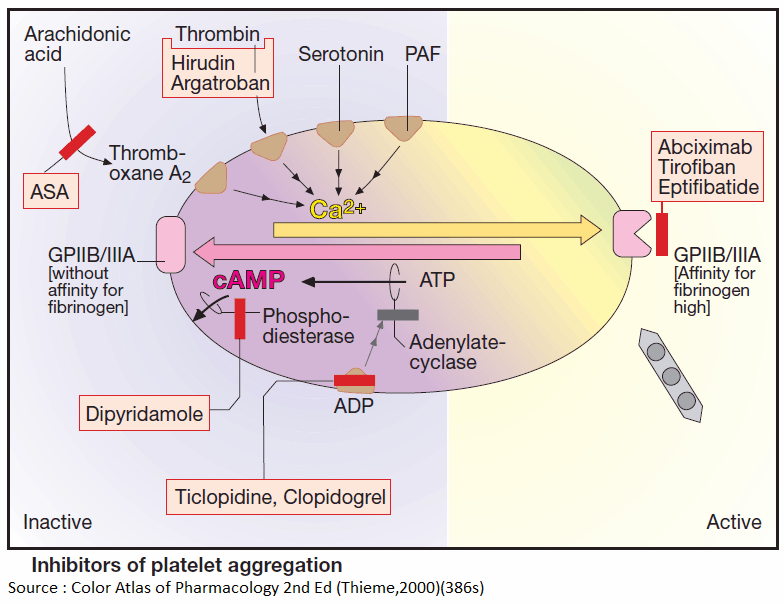
Acetylsalicylic Acid (ASA)
Platelet aggregation can be inhibited by acetylsalicylic acid (ASA), which blocks thromboxane synthase, or by recombinant hirudin (originally harvested from leech salivary gland), which binds and inactivates thrombin. As yet, no drugs are available for blocking aggregation induced by serotonin or PAF.
Presystemic Effect of Acetylsalicylic Acid
Inhibition of platelet aggregation by ASA is due to a selective blockade of platelet cyclooxygenase. Selectivity of this action results from acetylation of this enzyme during the initial passage of the platelets through splanchnic blood vessels. Acetylation of the enzyme is irreversible.
ASA present in the systemic circulation does not play a role in platelet inhibition. Since ASA undergoes extensive presystemic elimination, cyclooxygenases outside platelets, e.g., in endothelial cells, remain largely unaffected.
With regular intake, selectivity is enhanced further because the anuclear platelets are unable to resynthesize new enzyme and the inhibitory effects of consecutive doses are added to each other. However, in the endothelial cells, de novo synthesis of the enzyme permits restoration of prostacyclin production.
ADP Receptor Inhibitors
ADP-induced aggregation can be prevented by ticlopidine and clopidogrel; these agents are not classic receptor antagonists. ADP-induced aggregation is inhibited only in vivo but not in vitro in stored blood; moreover, once induced, inhibition is irreversible.
A possible explanation is that both agents already interfere with elements of ADP receptor signal transduction at the megakaryocytic stage. The ensuing functional defect would then be transmitted to newly formed platelets, which would be incapable of reversing it.
Prostacyclin (iloprost) and Dipyridamole
The intra-platelet levels of cAMP can be stabilized by prostacyclin or its analogues (e.g., iloprost) or by dipyridamole. The former activates adenyl cyclase via a G-protein-coupled receptor; the latter inhibits a phosphodiesterase that breaks down cAMP.
Glycoprotein IIB/IIIA Inhibitors
The integrin (GPIIB/IIIA)-antagonists prevent cross-linking of platelets. Their action is independent of the aggregation – inducing stimulus. Abciximab is a chimeric human-murine monoclonal antibody directed against GPIIb/IIIa that blocks the fibrinogen-binding site and thus prevents attachment of fibrinogen. The peptide derivatives, eptifibatide and tirofiban block GPIIB/IIIA competitively, more selectively and have a shorter effect than does abciximab.
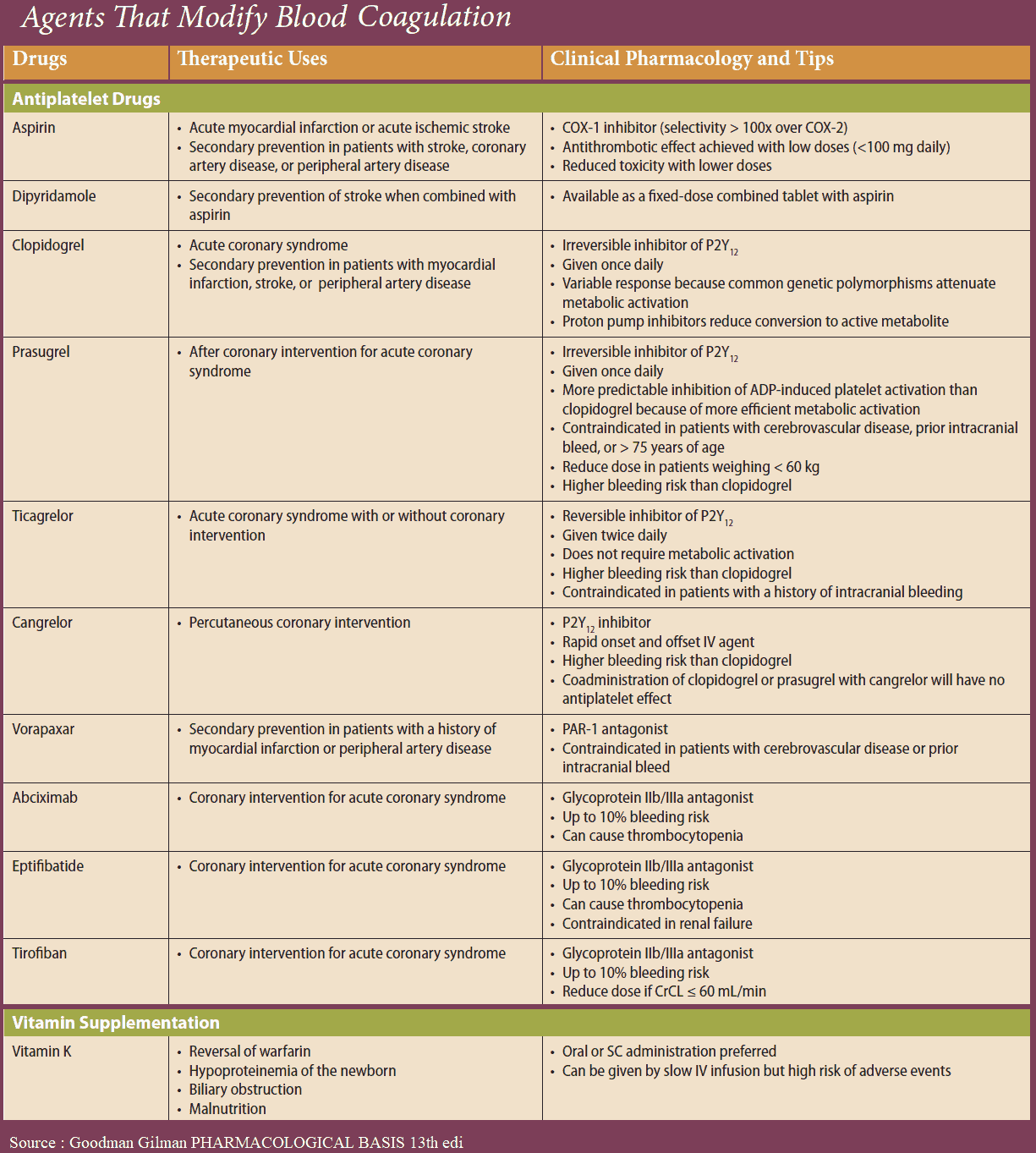
Adverse Effects of Antiplatelet Drugs
- All antiplatelet drugs increase the risk of bleeding.
- Even at the low ASA doses used to inhibit platelet function (100 mg/d), ulcerogenic and bronchoconstrictor (aspirin asthma) effects may occur.
- Ticlopidine frequently causes diarrhea and, more rarely, leukopenia, necessitating cessation of treatment. Clopidogrel reportedly does not cause hematological problems.
- As peptides, hirudin and abciximab need to be injected; therefore their use is restricted to intensive-care settings.
READ MORE about Anticoagulants and Fibrinolytics