This post is an answer to the Case – Patient with Right Subscapular Pain and Tachycardia
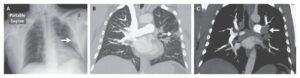
Chest radiograph was performed showing multiple right lung cysts associated with an increase in volume and the subsequent mediastinal shift to the left, but no pneumothorax.
Thoracic CT scan confirmed that the whole of the right lung was replaced with cysts (image below). Therefore, a presumptive diagnosis of congenital cystic adenomatoid malformation (CCAM) was made, which was then confirmed by histologic examination.
Differential Diagnosis
- Congenital cystic adenomatoid malformation, type I.
- Diaphragmal herniation
- Pulmonary sequestration
- Congenital lobar emphysema
- Bronchogenic cyst
Discussion
CCAM is a rare developmental, non-hereditary anomaly of the lung characterised by proliferation of terminal bronchiole-like interconnecting structures with formation of cysts of varying sizes. It constitutes up to 25% of congenital cystic lung lesions. It is classified into three subtypes based on the clinical, gross and histological features.
Type I (50–70% of cases) is characterised by multiple large cysts, (up to 10 cm) or a single dominant often multiloculated cyst surrounded by multiple smaller cysts. It has a good prognosis. Type II (20-40% of cases) shows multiple small cysts (< 2 cm). Type III is rare (10% of cases) and consists of a solid mass with cysts less than 0.5 cm mimicking the terminal bronchioles and the alveolar ducts; it is now considered a form of pulmonary hyperplasia. Types II and III have usually a poor prognosis because they are often associated with other congenital anomalies [1, 2].
The pathogenesis of CCAM is uncertain. The primary defect during development that leads to CCAM has been described as either bronchial atresia or maturation arrest in bronchopulmonary segments before the 17th week of gestation. The morphology of the lesion is then determined by the dysplastic lung growth beyond the atretic segment. In general, lungs with CCAM have a normal arterial supply and venous drainage. The malformation is usually unilateral and sublobar or lobar in size, but occasionally it can be multilobar or bilateral with no special predilection for any lobe.
Up to 70% of such cases are asymptomatic at birth, 80–85% of cases are diagnosed in the first 2 years of life with signs of respiratory distress; rarely the presentation is delayed until adulthood. Up to 25% of cases can be associated with other congenital anomalies, including extralobular sequestration, diaphragmatic hernia, pulmonary hypoplasia and cardiovascular malformations.
Clinically, adults most frequently show recurrent or persistent pulmonary infections. Other ways of presentation include haemoptysis, hemothorax, pneumothorax, pyopneumothorax, shortness of breath or as an incidental finding on a chest radiograph.The prenatal diagnosis can usually be established by ultrasonography. Chest X-ray and CT represent the postnatal diagnostic method of choice.
Radiological abnormalities range from a soft-tissue mass containing single or multiple air-filled cysts of varying sizes to a solid homogeneous mass. CT imaging has a good accuracy in characterising the various types and is indispensable in identifying the true extent of the lesion [3].
The differentiation of CCAM from the diaphragmatic hernia can be done noting the presence of abdominal contents in the chest cavity. Pulmonary sequestration appears as a well-circumscribed, solid mass that receives its blood from the aorta whereas CCAM is usually supplied by the pulmonary artery. Bronchogenic cysts are found predominantly near the carina, are small and appear as smooth round masses. Congenital lobar emphysema can be distinguished by the presence of bronchovascular markings extending to the periphery of the involved lobe and by atelectasis of adjacent tissues.
CCAM has been associated with the development of neoplasms (bronchioloalveolar carcinoma, blastomas and rhabdomyosarcoma). Treatment of CCAM is usually surgical, involving complete resection of the affected lung lobe [4].
References
- Khan NU, Jones MT, Greaves M (2008) Congenital cystic adenomatoid malformation of an entire lung in a 33-year-old man: a case report and review of the literature. The British Journal of Radiology 81: e276–e278 (PMID: 18941042)
- Morelli L, Piscioli I, Licci S, Donato S, Catalucci A, Del Nonno F (2007) Pulmonary congenital cystic adenomatoid malformation, type I, presenting as a single cyst of the middle lobe in an adult: case report. Diagnostic Pathology 2:17 (PMID: 17555585)
- Keidar S, Ben-Sira L, Weinberg M, Jaffa AJ, Silbiger A, Vinograd I (2001) The Postnatal Management of Congenital Cystic Adenomatoid Malformation. IMAJ 3:258-261 (PMID: 11344837)
- Mohta A, Kanojia RP, Bathla S, Khurana N (2009) Cystic adenomatoid malformation of the lung: A diagnostic dilemma. Afr J Paediatr Surg 6: 112-113 (PMID: 19661643)